Paired Pulsed Decarboxylative Hydroxylation Designed by Online Electrochemistry–Mass Spectrometry
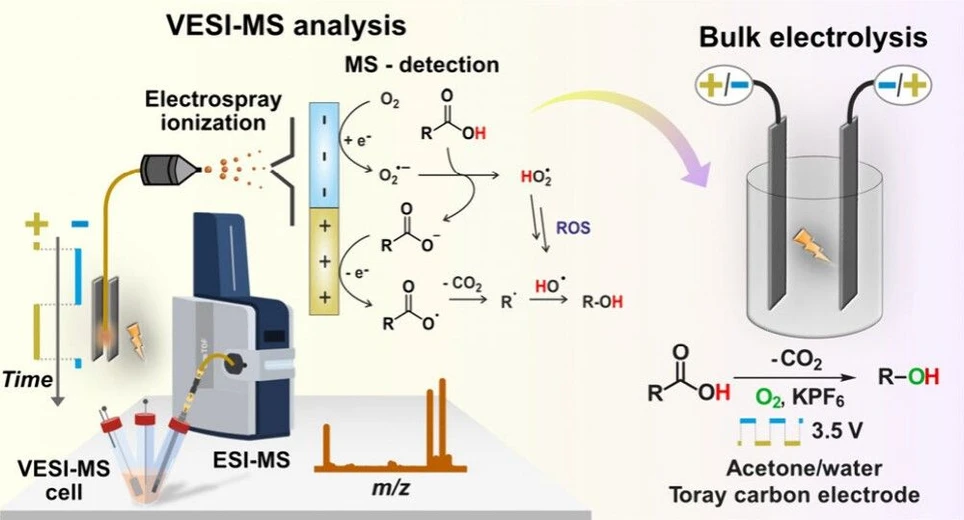
J. Am. Chem. Soc. 2025, 147, 41, 36975–36979: Graphical abstract
A new approach for optimizing paired pulsed electrosynthesis is demonstrated through real-time monitoring of electrode interfacial reactions using voltammetry–electrospray ionization mass spectrometry (VESI-MS). This technique provides molecular-level insight into redox events, enabling precise control and synchronization of oxidation and reduction pulses and supporting the discovery of new alternating current (AC) electrochemical transformations.
Applying this concept, we achieved a selective decarboxylative hydroxylation, coupling oxygen reduction with the oxidative decarboxylation of a carboxylic acid. In a pilot study with 2-phenylpropionic acid, the method produced 1-phenylethanol with excellent selectivity using only molecular oxygen, KPF₆, water, and carbon electrodes. The results establish VESI-MS as a powerful tool for rational design of pulsed electrochemical reactions.
The original article
Paired Pulsed Decarboxylative Hydroxylation Designed by Online Electrochemistry–Mass Spectrometry
Adarsh Koovakattil Surendran, and Jana Roithová*
J. Am. Chem. Soc. 2025, 147, 41, 36975–36979
https://doi.org/10.1021/jacs.5c12895
licensed under CC-BY 4.0
Selected sections from the article follow. Formats and hyperlinks were adapted from the original.
Electrosynthesis utilizes single or sequences of one-electron oxidation and reduction steps to enable clean chemical transformations that would typically require stoichiometric reagents, rare metal catalysts, or other activation methods. (1−8) However, this seemingly ideal approach is often complicated by undesired effects associated with the electrosynthesis process. For example, mass transport effects can lead to the accumulation of intermediates, triggering unwanted side reactions, and causing issues such as overoxidation, over-reduction, or electrode fouling due to uncontrolled interfacial conditions. (9−12) In many instances, effective chemical synthesis occurs at one electrode, while it is coupled with a sacrificial redox reaction at the other electrode (Figure 1a). (13,14)
 J. Am. Chem. Soc. 2025, 147, 41, 36975–36979: Figure 1. Examples of approaches to electrosynthesis. (a) DC Kolbe synthesis. (14,36) (b) rAP Kolbe synthesis. (14) (c) Convergent Paired Electrosynthesis. (22) (d) Paired Pulsed Electrosynthesis (developed here).
J. Am. Chem. Soc. 2025, 147, 41, 36975–36979: Figure 1. Examples of approaches to electrosynthesis. (a) DC Kolbe synthesis. (14,36) (b) rAP Kolbe synthesis. (14) (c) Convergent Paired Electrosynthesis. (22) (d) Paired Pulsed Electrosynthesis (developed here).
Recent advancements in the field have demonstrated that utilizing alternating current or alternating voltage pulses (AVP) can help reduce mass transport effects, local pH variation, and minimize electrode fouling (Figure 1b). (14−18) However, designing these pulses necessitates careful consideration of the chemistry at the electrode interface, which presents challenges that may hinder the development of AVP for electrosynthesis. (19−21)
Another promising development is the convergent pairing of electrochemical reactions, where both electrodes generate reactive intermediates that eventually couple to form a product (Figure 1c). (22−25) The generality of this approach is often hindered by the spatial separation of the initially formed reactive species, which frequently requires the formation of persistent radicals or other stabilized intermediates.
Electrochemistry – mass spectrometry couplings can provide molecular insight into electrochemical reactions. (26−31) We have recently developed a Voltammetry–Electrospray Ionization Mass Spectrometry (VESI-MS) approach to study chemical processes occurring near electrodes in real-time during the electrochemical reaction (Figures S1 and S2). (10,32) In this paper, we demonstrate how this approach can be employed to tune voltage pulses and design pulsed paired electrosynthesis. We specifically illustrate its application in the decarboxylative hydroxylation reaction (Figure 1d). By utilizing alternating voltage pulsing, we can selectively produce the alcohol while suppressing both radical dimerization and overoxidation. (33−36)
The hypothesis of the reaction posits that electrochemical oxygen reduction (ORR) and carboxylic acid oxidation (CAO) are complementary redox reactions. ORR is favored in acidic conditions, whereas CAO requires a basic environment. (14,36−38) The superoxide or peroxide species generated during the ORR can function as in situ bases, deprotonating carboxylic acids to produce carboxylates. (39,40) These carboxylates are then oxidized during the subsequent anodic pulse, leading to decarboxylation and the formation of alkyl radicals. (11,36,41) In the next step, the cathodic pulse reduces oxygen to hydroxyl radical or other reactive oxygen species (ROS), which may react with the alkyl radicals to form the corresponding alcohol. (42−44)
As oxidation and reduction alternate at each electrode, the same redox sequence occurs at both electrodes in tandem, eliminating the need for a sacrificial counter-reaction and enabling efficient paired electrolysis.
We tested our hypothesis with the VESI-MS experiments. First, we evaluated whether carboxylate can be generated during oxygen reduction on a Toray carbon electrode. We selected 2-phenylpropionic acid (RCOOH in the following) as the model substrate due to the stability of its secondary benzylic radical and cation. A linear sweep VESI-MS voltammogram from −0.2 to −1.95 V vs Pt, of a 2 mM RCOOH in acetone under O2 (0.12 bar) with KPF6 (2 mM) electrolyte confirms that carboxylate can be generated in situ during ORR (Figure 2a, see Figure S4 for the related mass spectra). The current caused by the oxygen reduction at the electrode correlates with the depletion of the carboxylic acid signal ([RCOOH]K+) and a simultaneous rise of the signal of carboxylate anions ([RCOO–]2K+). We use KPF6 rather than the commonly used tetrabutylammonium salts because it is cheaper and more compatible with ESI-MS measurements. In addition, potassium ions often bind to neutral molecules during the ionization, enabling their detection by ESI-MS, as observed here.
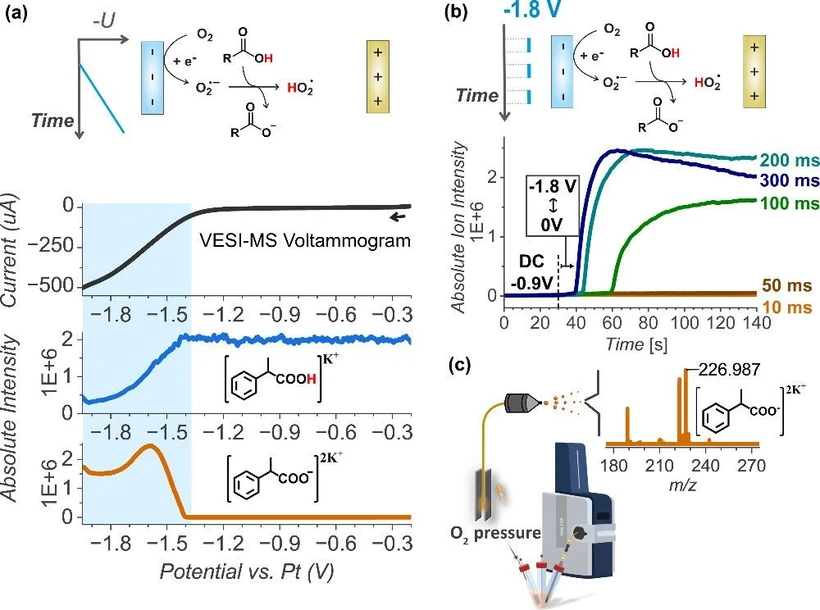 J. Am. Chem. Soc. 2025, 147, 41, 36975–36979: Figure 2. (a) Proof of concept: oxygen reduction at the electrode leads to carboxylic acid deprotonation. (b) Tuning the reduction pulse length for the maximum carboxylate formation. (c) VESI-MS setup.
J. Am. Chem. Soc. 2025, 147, 41, 36975–36979: Figure 2. (a) Proof of concept: oxygen reduction at the electrode leads to carboxylic acid deprotonation. (b) Tuning the reduction pulse length for the maximum carboxylate formation. (c) VESI-MS setup.
To effectively couple the oxygen reduction reaction (ORR) with carboxylate oxidation, we need to estimate the time scales of each reaction: the duration of the reduction pulse for maximizing carboxylate formation and the optimal oxidation pulse length for complete decarboxylation. We focused first on optimizing the reduction pulse. We applied symmetric square wave pulses between −1.8 V (vs Pt) (where ORR and carboxylate formation occur) and 0 V (where no reaction takes place). Pulse durations were varied from 100 μs to 300 ms (100 μs, 1, 10, 50, 100, 200, and 300 ms), and the carboxylate formation was monitored in real-time with mass spectrometry. To establish a nonpulsed baseline, the electrode was held at −0.9 V (vs Pt) for 30 s, followed by pulsing for about 2 min to ensure adequate detection time. The transfer time between the electrode and ESI-MS detection was about 10 s. No carboxylate signal was observed for pulse durations below 10 ms, while a slight increase was seen at 10 ms, followed by a sharp rise at 100 ms, reaching a maximum at 200–300 ms (Figure 2b and Figure S5). The onset of the carboxylate signal is influenced by its concentration; lower concentrations require longer times for detectable accumulation. Hence, a 300 ms pulse maximizes the concentration of carboxylate, but we reach the diffusion limit since the same amount of carboxylate can be produced with 200 ms pulses over a longer period.
We chose to fix the 200 ms reduction pulse duration at −1.8 V and proceeded with optimizing the oxidation pulse. The oxidation voltage was set to 1.7 V (vs Pt) as determined from VESI-MS experiments with a constant −1.8 V (vs Pt) (200 ms) reduction pulse and systematically increasing the oxidation pulse potential from 1 to 1.7 V (vs Pt) (Figure S6, the pulse duration was symmetrical, 200 ms). At 1.7 V (vs Pt), all the carboxylate ions generated from the reduction pulse are oxidized, leading to the disappearance of the [RCOO–]2K+ ions from the mass spectra. Next, we varied the oxidation pulse duration, keeping the offset of 1.7 V (vs Pt), from 100 μs to 600 ms, while monitoring the depletion of the carboxylate signal (Figure 3a and Figure S7, the reduction pulses were kept constant at −1.8 V (vs Pt) for 200 ms). The experimental sequence again started by holding the average DC potential (−0.05 V) for 30 s, followed by recording the effects of pulsing for 2 min.
 J. Am. Chem. Soc. 2025, 147, 41, 36975–36979: Figure 3. Pairing the 200 ms reduction pulses with the oxidation pulses.
J. Am. Chem. Soc. 2025, 147, 41, 36975–36979: Figure 3. Pairing the 200 ms reduction pulses with the oxidation pulses.