Automated Mag-Net Enrichment Unlocks Deep and Cost-Effective LC–MS Plasma Proteomics
J. Proteome Res. 2026, 25, 2, 618–632: Graphical abstract
Plasma proteomics is challenged by high-abundance proteins that mask low-abundance biomarkers. This study compares five enrichment strategies and identifies Mag-Net as a scalable and cost-effective approach, achieving over 4200 protein identifications per sample using short LC gradients and data-independent acquisition.
Further optimization and automation of Mag-Net on a liquid handling platform, combined with advanced mass spectrometry, increased coverage to 4500 proteins per sample with a throughput of 100 samples per day. The workflow demonstrated high reproducibility and minimal per-sample cost, providing an efficient solution for large-cohort biomarker discovery in plasma proteomics.
The original article
Automated Mag-Net Enrichment Unlocks Deep and Cost-Effective LC–MS Plasma Proteomics
Erkka Järvinen, Xiaonan Liu, Markku Varjosalo, and Salla Keskitalo*
J. Proteome Res. 2026, 25, 2, 618–632
https://doi.org/10.1021/acs.jproteome.5c00420
licensed under CC-BY 4.0
Selected sections from the article follow. Formats and hyperlinks were adapted from the original.
Typical liquid chromatography–mass spectrometry (LC–MS)-based untargeted proteomics of neat plasma identifies around 500 proteins, as most peptides detected by LC–MS are derived from a few hundred highly abundant proteins, hindering the detection of low-abundant proteins. (4−6) Still, plasma contains large numbers of cellular leakage proteins that are present at significantly lower concentrations than classical plasma proteins. (1−3,7) Several techniques have been developed to improve protein identifications from neat plasma, including immunodepletion of abundant proteins combined with or without peptide prefractionation, and various solvent- or acid–based precipitation workflows. (4,7−9) However, these methods typically reach no more than ∼1000 protein identifications. (7−9)
Targeted antibody- or aptamer-based affinity proteomics methods, which detect predefined panels of proteins, have gained popularity as alternatives to untargeted LC–MS proteomics. (4) A recent publication profiled the plasma proteome of over 50,000 adults using an early generation antibody assay measuring up to 3000 proteins. (10) The latest version of the aptamer-based assay reportedly covers nearly 10,000 unique protein targets. (11) A key drawback, however, is that antibodies and aptamers may be affected by epitope variations due to genetic polymorphisms or post-translational modifications, and may suffer from cross-reactivity between targets and thus limiting their specificity compared to LC–MS. (4,12)
Plasma protein enrichment methods have recently expanded protein identifications up to 5000 plasma proteins. These methods include nanoparticles that form protein coronas when mixed with plasma (e.g., Proteograph Assay), (13,14) hyper-porous magnetic beads with quaternary ammonium groups that bind to phospholipid bilayer-bound particles in (Mag-Net), (15,16) and other commercial magnetic particle-based enrichment methods (e.g., ENRICHiST, ENRICHplus, Proteonano). (14,17,18) With Lycopersicon esculentum lectin-mediated plasma enrichment, up to 8500 proteins were reported to be detected from just 25 μL of plasma. (19) Similarly, extracellular vesicles (EVs) and other extracellular particles carrying tissue-derived proteins have emerged as promising targets to increase plasma proteome depth. (20−23)
Extracting EVs and other extracellular particles from plasma remains a key bottleneck for large scale studies. (24) Traditional methods such as differential centrifugation or size exclusion chromatography are labor-intensive and difficult to scale for large plasma cohorts. Magnetic bead-based isolation of plasma EVs, by capturing phosphatidylserine or CD9+CD63+CD81 positive EVs, have been proposed as a scalable alternative. (23,25) These approaches have shown comparable or better performance than differential centrifugation, enabling the identification of more than 4,000 proteins from human plasma. (5,22,23,25,26) Moreover, magnetic bead-based methods are compatible with automation, making them especially attractive for high-throughput plasma proteomics.
The potential of LC–MS proteomics using data-independent acquisition (DIA) has been demonstrated in recent studies analyzing cohorts ranging from several hundred to thousands of plasma samples, achieving throughput rates of up to 60 samples per day. (8,27−29) To analyze such large cohorts, automation of sample preparation workflows is essential to reduce manual intervention and improve reproducibility. Fully automated workflows now exist that integrate all key steps of plasma proteomics, including protein extraction, reduction, alkylation, digestion, and peptide loading into disposable trap columns (Evotips), preparing samples directly for LC–MS analysis. (6) Some systems even support solid-phase peptide purification within the liquid handling platform. (28)
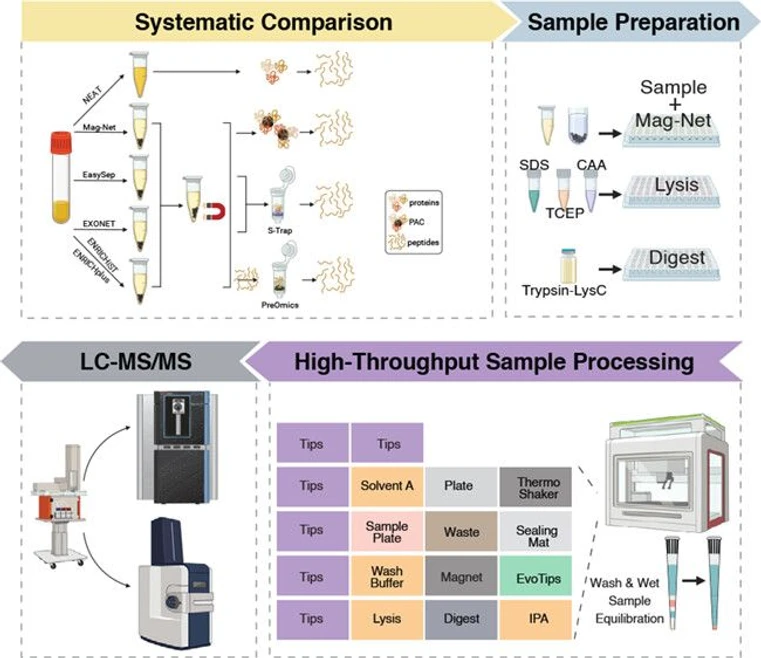
In this study, we systematically evaluated five different plasma enrichment methods, alongside neat plasma, and benchmarked their performance in LC–MS proteomics. We selected the Mag-Net workflow, (15,16) two commercial enrichment kits (ENRICHiST and ENRICHplus), and two commercial antibody-based EV enrichment beads (EasySep and EXONET). We also assessed the effect of LC gradient length on protein and precursor identifications. Finally, we implemented the Mag-Net workflow on a liquid handling workstation, streamlining the process from plasma enrichment to Evotip loading for LC–MS-ready samples. This work establishes a scalable, cost-effective framework for high-throughput plasma proteomics, enabling the analysis of thousands of plasma samples in large-scale studies.
Materials and Methods
LC–MS/MS
Peptide samples were loaded into Evotips according to the manufacturer’s protocol by conditioning tips with 20 μL of 0.1% formic acid in acetonitrile followed by soaking them in isopropanol. The tips were equilibrated with 20 μL of solvent A before loading 10 to 30 μL of samples. The tips were further washed with 20 μL of solvent A before briefly centrifuging them with 100 μL of solvent A and leaving the rest of the solvent A in the tips. Sample loads were 100, 200, and 300 ng of peptides for 100 samples per day (SPD), 60 SPD, and 30 SPD methods, respectively. For neat plasma analysis, sample loads were 10, 20, and 30 μL for the 100 SPD, 60 SPD and 30 SPD methods, respectively. The Evosep One liquid chromatography (LC) system (33) connected to a trapped ion mobility quadrupole time-of-flight MS (Bruker timsTOF Pro 2, Bruker Daltonics, Billerica, MA, US) equipped with CaptiveSpray nanoelectrospray ion source (Bruker Daltonics) was employed for the LC–MS analysis. A 15 cm × 150 μm column with 1.5 μm C18 beads (EV1137, Evosep) was employed for the 30 SPD (44 min gradient) method. An 8 cm × 150 μm column with 1.5 μm C18 beads (EV1109, Evosep) was employed for the 60 SPD (21 min gradient) and 100 SPD (11.5 min gradient) methods. The column oven was set to a temperature of 40 °C for all methods. Mobile phases A and B were 0.1% formic acid in water and 0.1% formic acid in acetonitrile. For the MS data acquisition, DIA with parallel accumulation-serial fragmentation (dia-PASEF) (34) was used with default source parameters and optimized isolation windows (Tables S2–S6).
For the automated Mag-Net samples, the Orbitrap Astral MS controlled with Thermo Tune software (version 1.1.477.46), both from Thermo Fisher Scientific, and coupled to an Evosep One was employed. The samples were analyzed with the 100 SPD method using an 8 cm x 150 μm Aurora Rapid XT analytical column from IonOpticks (Collingwood, Victoria, Australia) interfaced online using an EASY-Spray source from Thermo Fisher Scientific. The Orbitrap Astral MS was operated at a full MS resolution of 240,000 with a full scan range of 380–980 m/z. RF lens was set to 40%, and the full-MS AGC target was set to 500%. Maximum injection time for full-MS was 5 ms. MS/MS scans for DIA analysis were recorded with 3 Th isolation window from 380 to 980 m/z, 7 ms maximum ion injection time and AGC target 500%. The isolated ions were fragmented using HCD with 25% normalized collision energy.
Results
Automated Plasma Enrichment Workflow
We selected the Mag-Net workflow for automation due to its significantly lower cost - over an order of magnitude cheaper than the other enrichment methods (Table S8). Furthermore, it is only workflow that does not require cartridge-based purification. Therefore, we developed an automated plasma enrichment method for the Biomek i5 liquid handler based on the Mag-Net workflow, fully integrating it with a novel, automated Evotip loading method. First, we confirmed that the automated Evotip loading (Figure S12A) performed comparably to manual Evotip loading (Figure S12B).
To evaluate the performance of automated Mag-Net workflow, we processed two 96-well plates, each containing 15 replicates of six plasma samples, and 6 replicates of blank samples without plasma (Figure 4A). In addition, we included six QC samples for the LC–MS analysis of each plate, prepared by manually loading 50 ng of HeLa protein digest standard into Evotips. Finally, to assess the consistency across different mass spectrometry platforms, we analyzed the same sample plates using both the Orbitrap Astral (Figure 4) and the timsTOF Pro 2 (Figure S12).
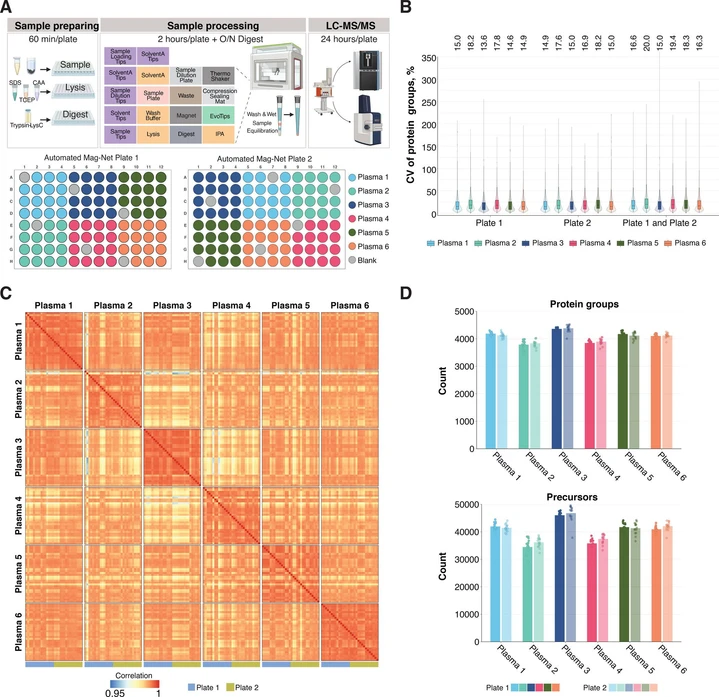 J. Proteome Res. 2026, 25, 2, 618–632: Figure 4. Automated Mag-Net workflow with combined Evotip loading using a Biomek i5 liquid handler and analyzed with an Orbitrap Astral mass spectrometer shows high reproducibility. (A) Overview of the automated Mag-Net and Evotip workflows and plate layouts for two Mag-Net runs. The 100 SPD method was used for LC–MS analysis of all samples. (B) Coefficient of variation (CV, %) of protein groups within each plasma sample across plate 1 and plate 2 (n = 15 per plate; n = 30 total). The exact values of median CVs are shown above the plot. (C) Pearson correlation of protein intensities between plasma samples across both plates. The scale bar is from 0.9 to 1. (D) Number of identified protein groups and precursors per plasma sample on each plate (n = 15 per plate; n = 30 total).
J. Proteome Res. 2026, 25, 2, 618–632: Figure 4. Automated Mag-Net workflow with combined Evotip loading using a Biomek i5 liquid handler and analyzed with an Orbitrap Astral mass spectrometer shows high reproducibility. (A) Overview of the automated Mag-Net and Evotip workflows and plate layouts for two Mag-Net runs. The 100 SPD method was used for LC–MS analysis of all samples. (B) Coefficient of variation (CV, %) of protein groups within each plasma sample across plate 1 and plate 2 (n = 15 per plate; n = 30 total). The exact values of median CVs are shown above the plot. (C) Pearson correlation of protein intensities between plasma samples across both plates. The scale bar is from 0.9 to 1. (D) Number of identified protein groups and precursors per plasma sample on each plate (n = 15 per plate; n = 30 total).
The QC samples demonstrated consistent performance across both batches and instruments (Figures S12B and S13A) indicating no bias toward a specific MS platform. The coefficient of variation (CV) of protein quantification across the full Mag-Net workflow, from plasma sample to quantified protein values, was well below or at 20% within and between plates in both instruments (Figures 4B and S12C). The CVs of precursor quantification were higher and fell between 20 and 30% (Figures S12D and S13B). Although some proteins were identified in blank samples, their total precursor counts never exceeded 0.7% of average sample count in either of instruments.
The correlation of protein quantification within the same plasma samples was mostly 0.99 for the Orbitrap Astral, and no differences were observed between the plates indicating the absence of batch effects or well position biases (Figure 4C). Similarly, for samples analyzed using the timsTOF Pro 2, we observed excellent correlation across replicates, although the absolute values were slightly lower (Figure S12E).
MS1 signal intensity remained largely stable, except for the first ten runs of Plate 2, which showed slightly elevated intensity compared to Plate 1 and the remaining samples in both instruments (Figures S12G and S13C). However, this variation did not affect sample correlation or protein and precursor identifications (Figures 4C,D, and S12E,F). The elevated MS1 signal was likely due to a slight plate edge effect, as the wells in row A of Plate 2 appeared to contain reduced liquid volume, despite proper sealing the plate with foil.
The total number of protein groups and precursors identified was highly consistent across replicates (Figures 4D and S12F). As expected, the Orbitrap Astral identified twice as many proteins and three times as many precursors as the timsTOF Pro 2, despite using the same LC gradient (Figures 4D and S12F). A direct comparison between the manual (Figure 1C) and automated workflows (Figure S12F), performed on the same instrument with the identical LC gradient, confirms that both workflows yield comparable protein identification results from plasma. Finally, we evaluated the digestion efficiency of the automated Mag-Net workflow, as this step initially required multiple rounds of optimization. Digestion efficiency was consistent across all samples and both plates, with a mean value of 16% and a range from 14 to 18% (Figure S12H). For comparison, the manual Mag-Net workflow yielded an average digestion efficiency of 14% (n = 9).
Discussion
Increased throughput in LC–MS analysis also demands higher throughput for sample preparation. Here, we developed an automated version of Mag-Net in a 96-well plate format for the Biomek i5 liquid handler. The workflow fully integrates all steps, starting from plasma sample to LC–MS ready samples loaded in the disposable Evotip trap columns. A similar workflow has previously been reported for the magnetic bead protein aggregation capture workflow integrated with Evotip loading in the Opentrons OT-2 liquid handling robot. (6) Our Mag-Net method demonstrated excellent reproducibility, with an average CV of 18% for protein quantification across two full 96-well plates and six different plasma samples. This result is highly comparable to what has been previously reported for the manual Mag-Net workflow (13 and 21%) (15,18) or for a semiautomated acid precipitation method for neat plasma (16%), (8) and better than reported for an automated neat plasma workflow (33%). (29) The development of the automated Mag-Net workflow relied on the high binding capacity and rapid magnetization-based recovery of SAX beads, allowing us to miniaturize the bead volume per well to 0.5 μL while simultaneously reducing the amount of digestion enzyme to 90 ng per well. Even after miniaturizing the bead volume, we had to dilute the samples 5- to 10-fold before loading them into Evotips to achieve suitable input for the mass spectrometers. Another factor for further optimization is the digestion step to reduce the time required for sample preparation. Most of the time in the current workflow is spent on the overnight digestion at 37 °C. It has been shown that on-bead digestion can be conducted efficiently even within 4 h at 37 °C. (6) Finally, the current automated method could be readily scaled up to process six plates in a single run, depending on the capacity of the liquid handler.
In conclusion, we compared five different plasma protein enrichment methods to neat plasma. The four best-performing enrichment methods (Mag-Net, ENRICHplus, EasySep and EXONET) increased protein identification 5- to 7-fold in comparison to neat plasma. The highest number of proteins identified from a single plasma sample exceeded 4000 proteins when using a 30 SPD chromatographic method. We further developed a fully automated plasma enrichment method for 20 μL of plasma using the Mag-Net method. This method offers exceptionally low total sample cost of less than 4$, alongside excellent reproducibility with a coefficient of variation below 20% for protein quantification. It also enables outstanding protein identification, detecting approximately 4000 proteins from a single plasma sample with a throughput of one 96-well plate per day.