Comparative Evaluation of Total Ergot Alkaloids Determination by ELISA and UHPLC-MS/MS in Wheat: Implications of Cross-Reactivity and Matrix Effects
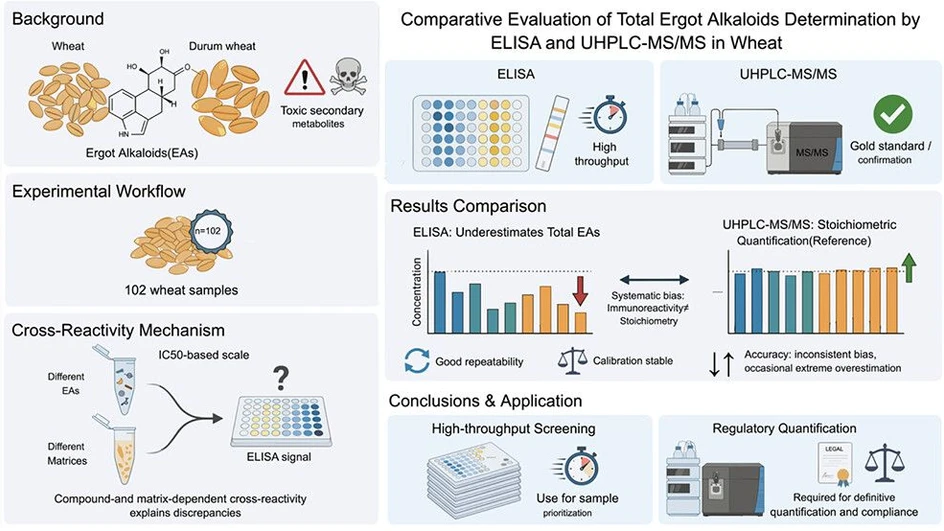
J. Agric. Food Chem. 2026, 74, 25, 20015–20027: Graphical abstract
This study compares a commercial ELISA assay with a validated UHPLC-MS/MS method for determining total ergot alkaloids in wheat and durum samples. In addition to method performance, the authors investigated cross-reactivity and matrix effects to explain differences between immunoassay and chromatographic results.
While ELISA demonstrated good repeatability and is well suited for high-throughput screening, systematic biases were observed due to compound- and matrix-dependent cross-reactivity. The findings support ELISA as an effective screening tool, whereas UHPLC-MS/MS remains the preferred method for accurate quantification and regulatory confirmation of ergot alkaloids.
The original article
Comparative Evaluation of Total Ergot Alkaloids Determination by ELISA and UHPLC-MS/MS in Wheat: Implications of Cross-Reactivity and Matrix Effects
Chamali Kodikara*, Dainna Drul, Nandika Bandara, and Sheryl A. Tittlemier*
J. Agric. Food Chem. 2026, 74, 25, 20015–20027
https://doi.org/10.1021/acs.jafc.6c05234
licensed under CC-BY 4.0
Selected sections from the article follow. Formats and hyperlinks were adapted from the original.
Ergot alkaloids (EAs) are a structurally diverse group of indole-derived mycotoxins produced primarily by phytopathogenic fungi of the genus Claviceps, most notably Claviceps purpurea. These fungi infect the flowering heads of cereal crops, replacing the developing grain with hardened fungal structures (sclerotia) that contain ergot alkaloids. Cereals such as rye, wheat, barley, oats, and triticale are particularly susceptible, with infection severity strongly influenced by environmental conditions during flowering, including rainfall and humidity. (1−3) Although improvements in agronomic practices and grain cleaning have reduced the prevalence of visible ergot sclerotia, ergot alkaloids can persist in cereals and cereal-based foods even in the absence of detectable sclerotia. This may occur because sclerotia can fragment during harvesting, handling, cleaning, or milling, producing small particles that are not visually detected but may still contribute ergot alkaloids to grain or processed products, continuing to pose food and feed safety concerns. (4) (5) Ergot alkaloids share a common tetracyclic ergoline backbone and occur as structurally related compounds, including ergometrine, ergotamine, ergosine, ergocristine, ergocryptine, and ergocornine, together with their corresponding C-8 epimers. These epimers occur as R- and S-configurations, commonly referred to as -ine and -inine forms, respectively. Epimerization at the C-8 position can occur during fungal metabolism and during sample extraction, storage, and analysis, complicating accurate quantification. (6,7) Although S-epimers were historically considered biologically inactive, recent evidence indicates that they may contribute to overall exposure and toxicity, supporting their inclusion in analytical and regulatory frameworks. (3)
Dietary exposure to elevated levels of ergot alkaloids has been associated with a range of adverse health effects, collectively referred to as ergotism, including vasoconstriction, ischemia, neurological disturbances, and hallucinations. (3) These effects arise primarily from interactions with adrenergic, serotonergic, and dopaminergic receptors. (8−10) In response, regulatory limits for ergot alkaloids in food have been introduced or revised in several jurisdictions. In the European Union, maximum levels have been established for the sum of 12 ergot alkaloids in cereal milling products under Regulation (EU) 2021/1399, which was subsequently consolidated under Regulation (EU) 2023/915. This sum includes ergometrine, ergosine, ergotamine, ergocornine, ergocryptine, and ergocristine together with their corresponding -inine epimers, increasing the demand for reliable analytical methods to support routine monitoring and enforcement. (1,11)
Liquid chromatography-tandem mass spectrometry (LC-MS/MS), particularly ultrahigh performance liquid chromatography-tandem mass spectrometry (UHPLC-MS/MS), is widely regarded as the reference technique for confirmatory determination of ergot alkaloids due to its sensitivity, selectivity, and ability to resolve epimeric forms. Recent methods routinely achieve low microgram-per-kilogram limits of quantification across a range of cereal matrices. (12−17) However, LC-MS/MS approaches are resource-intensive and typically confined to centralized laboratories, limiting their suitability for high-throughput screening or rapid decision-making in grain-handling and quality-control settings. Consequently, rapid screening methods, most notably enzyme-linked immunosorbent assays (ELISAs), are commonly employed as complementary tools to chromatographic analysis. (18−21) ELISAs offer reduced analysis time, simplified workflows, and lower per-sample costs, making them attractive for preliminary screening and sample triage. (22−24)
In contrast to other regulated mycotoxins, however, the commercial availability of ELISAs for ergot alkaloids is limited. While multiple kits have been evaluated, performance has varied substantially across assays and matrices. (1) At present, the Randox Food Diagnostics Ergot Alkaloids ELISA is the only widely marketed microplate ELISA that quantifies the sum of 12 major ergot alkaloids, comprising ergometrine, ergosine, ergotamine, ergocornine, ergocryptine, and ergocristine together with their corresponding -inine epimers, consistent with the analyte scope used for EU maximum levels for ergot alkaloids in specified cereal products. (25)
In addition, cereal matrix effects may influence antibody binding and signal generation, potentially resulting in biased quantification relative to LC-MS/MS reference methods. These limitations are particularly relevant given the compositional variability of ergot alkaloids across cereal species, geographic regions, and growing conditions. (12) Recent reviews emphasize the need for systematic, matrix-specific evaluation of immunoassay performance before such methods can be confidently integrated into regulatory or industrial monitoring programs. (1,6,11) However, independent evaluations of commercially available ergot alkaloid ELISAs, particularly those benchmarked against validated UHPLC-MS/MS methods using naturally infested wheat samples, remain limited.
In this context, we hypothesized that measurements of total ergot alkaloids obtained using a commercially available ELISA are influenced by matrix effects and antibody cross-reactivity, resulting in systematic differences compared to UHPLC-MS/MS, which provides compound-specific quantification. To test this, the objective of the present study was to conduct a comprehensive performance evaluation of a commercially available ELISA for the detection of ergot alkaloids in wheat, using UHPLC-MS/MS as a reference method. Specific aims included assessment of repeatability and intermediate precision, extraction efficiency and extract stability, the influence of test portion mass, method accuracy relative to UHPLC-MS/MS, and characterization of antibody cross-reactivity toward individual ergot alkaloids in both solvent and wheat matrix. The results provide data-driven guidance on the appropriate use of ELISA-based screening within tiered analytical strategies for regulatory monitoring and food safety decision-making.
2. Materials and Methods
2.3. UHPLC-MS/MS Reference Method
UHPLC-MS/MS analysis of ergot alkaloids was performed using the validated Canadian Grain Commission (CGC) method described by Tittlemier et al. (2015). Briefly, ground grain (10 g) was extracted with 50 mL of acetonitrile/aqueous ammonium carbonate (84:16, v/v; 3.03 mM ammonium carbonate) to provide alkaline extraction conditions, then shaken for 30 min and centrifuged at 2450g for 10 min at room temperature. An aliquot of extract (464 μL) was combined with internal standard solution (dihydroergotamine; 20 μL of 4 ng/μL) and aqueous ammonium carbonate (1536 μL), vortex-mixed, and filtered through a 0.45 μm polytetrafluoroethylene syringe filter before injection into the UHPLC-MS/MS system. As in the validated reference method, dihydroergotamine was added after extraction to correct for postextraction analytical variability; extraction recovery was evaluated during method validation. Chromatographic separation was achieved using a reversed-phase BEH C18 column (2.1 × 100 mm, 1.7 μm; Waters, Milford, MA, USA) with the autosampler maintained at 15 °C and the column at 30 °C. Mobile phases consisted of (A) 3.03 mM aqueous ammonium carbonate and (B) acetonitrile. The gradient program was as follows: initial, 75% A/25% B; 1.00 min, 60% A/40% B; 5.00 min, 40% A/60% B; 8.00 min, 22% A/78% B; 10.50–11.00 min, 10% A/90% B; and 13.00–16.00 min, 75% A/25% B, with a total run time of 16 min. Detection was performed using positive electrospray ionization in multiple reaction monitoring mode on a Waters Xevo triple quadrupole mass spectrometer (Waters, Milford, MA, USA). Under the validated chromatographic conditions, dihydroergotamine did not coelute with ergotamine, ergotaminine, or the other target ergot alkaloids. Identification and quantification were based on retention-time criteria and compound-specific MRM transitions, thereby minimizing potential isotopic or coelution interference. Precautions to minimize epimerization, including controlled handling of standards and extracts and reduced light exposure, were followed as described in the reference method. UHPLC-MS/MS quantification was performed using analyte-specific, matrix-matched calibration curves prepared according to the validated CGC reference method, with calibration levels of 0.046, 0.2, 1, 2, 10, and 20 μg/kg. Dihydroergotamine was used as the internal standard, and concentrations were calculated from internal-standard-normalized analyte responses with method dilution factors applied. Total ergot alkaloid concentration was calculated as the arithmetic sum of all quantified ergot alkaloids and epimers included in the validated method. (26)
3. Results and Discussion
3.4. Comparison of ELISA and UHPLC-MS/MS Results
For method comparison, UHPLC-MS/MS was used as the chemical reference method, providing compound-specific quantification of individual ergot alkaloids and their epimers. A total ergot alkaloid concentration was calculated as the arithmetic sum of all quantified alkaloids included in the method. This summed value represents a chemically defined total based on compound-specific quantification of each analyte, rather than an immunoreactivity-weighted response governed by antibody affinity. As such, the UHPLC-MS/MS total serves as a chemically defined comparator against which the immunoreactivity-weighted ELISA response can be interpreted. Agreement between the commercial ELISA and the validated UHPLC-MS/MS reference method was evaluated using wheat and durum wheat cargo samples (n = 102) collected over multiple crop years (Figure 5). Linear regression analysis demonstrated a positive association between ELISA and UHPLC-MS/MS results across the full concentration range, indicating that the ELISA was responsive to increasing ergot alkaloid levels. However, the regression slope deviated substantially from unity, with ELISA results systematically lower than UHPLC-MS/MS values, indicating an approximate 2-fold underestimation.
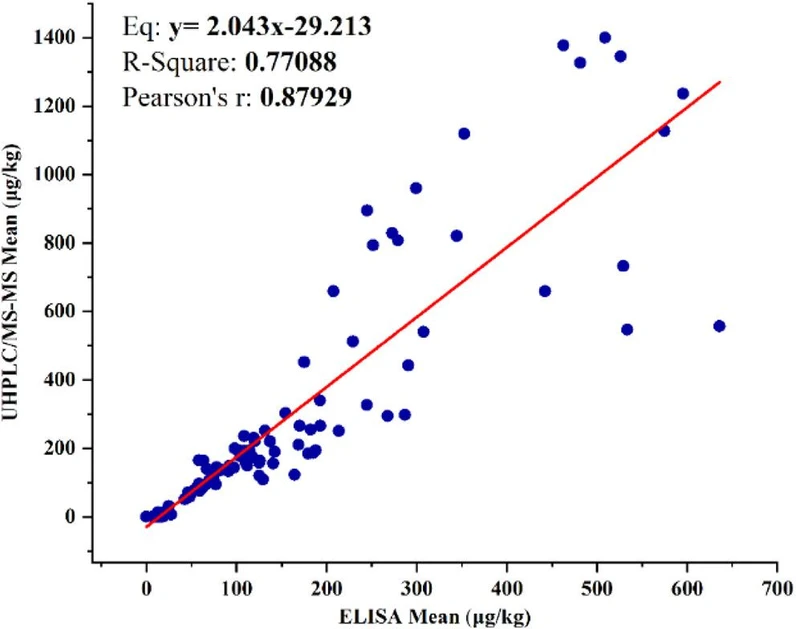 J. Agric. Food Chem. 2026, 74, 25, 20015–20027: Figure 5. Linear regression analysis comparing total ergot alkaloid concentrations determined by ELISA and UHPLC-MS/MS for wheat and durum wheat cargo samples (n = 102). Each point represents the mean of duplicate measurements (extracted from the same test portion) ELISA determinations for an individual cargo sample, plotted against the corresponding UHPLC-MS/MS result. Statistical analysis was performed using linear regression.
J. Agric. Food Chem. 2026, 74, 25, 20015–20027: Figure 5. Linear regression analysis comparing total ergot alkaloid concentrations determined by ELISA and UHPLC-MS/MS for wheat and durum wheat cargo samples (n = 102). Each point represents the mean of duplicate measurements (extracted from the same test portion) ELISA determinations for an individual cargo sample, plotted against the corresponding UHPLC-MS/MS result. Statistical analysis was performed using linear regression.
Bland-Altman analysis confirmed this proportional bias, showing a consistent negative mean difference between ELISA and UHPLC-MS/MS results across the concentration range (Figure 6). Importantly, the magnitude of disagreement did not increase randomly with concentration; rather, it reflected a systematic offset, indicating that the discrepancy was not driven by analytical imprecision or outliers but by fundamental differences in method response characteristics. This pattern indicates method-level bias rather than random analytical variability, which may arise from combined workflow effects including extraction, matrix response, and immunochemical cross-reactivity. The magnitude of the ELISA-UHPLC-MS/MS bias was substantially greater than the intermediate precision observed for the ELISA quality-control material, supporting the interpretation that the disagreement reflects systematic differences in method response rather than insufficient ELISA repeatability. Such behavior is well documented when immunoassays are compared with chromatographic methods for complex mycotoxin mixtures, including ergot alkaloids and other structurally diverse mycotoxins in cereals and feed. (28,30,33) These studies consistently report matrix- and profile-dependent bias in ELISA relative to LC-MS/MS, arising from differential antibody cross-reactivity and the summed immunoreactivity response of the assay, because ELISA produces an immunoreactivity-weighted total that depends on antibody affinity toward individual alkaloids and their relative abundance in the sample, whereas UHPLC-MS/MS quantifies compound-specific concentrations of each alkaloid and its epimers using compound-specific transitions and response factors, and total ergot alkaloid concentration is calculated as the sum of these targeted analytes. In contrast, ELISA produces an immunoreactivity-weighted total, determined by antibody affinity toward individual alkaloids and their competitive binding behavior within the assay format. Consequently, agreement between ELISA and UHPLC-MS/MS depends not only on total concentration, but also on the relative distribution of individual alkaloids within each sample. Although differences in extraction solvent composition and pH may influence ergot alkaloid recovery, stability, and epimeric distribution, each method was applied according to its intended analytical workflow. The ELISA was performed using the manufacturer-recommended acidic methanol/water extraction, whereas UHPLC-MS/MS was performed using the validated acetonitrile/aqueous ammonium carbonate extraction procedure. Therefore, the comparison reflects the performance of the complete methods as used in practice, rather than an isolated comparison of extraction solvents. The sequential extraction experiment further showed that repeating the ELISA extraction under the same kit conditions did not substantially increase the ELISA-measured response, supporting the adequacy of the kit extraction within the ELISA workflow while recognizing that it does not directly compare acidic and ammonium carbonate-based extraction conditions. Previous ELISA-LC-MS/MS comparison studies for ergot alkaloids have reported both under- and overestimation depending on assay design and matrix, including differences in alkaloid profile, cereal type, and antibody cross-reactivity. Turner et al. observed substantial overestimation of ergot alkaloids by ELISA relative to HPLC in rye field samples, attributed to strong antibody cross-reactivity toward alkaloids prevalent in rye matrices. Similar profile-dependent bias has also been reported in interlaboratory and method-comparison studies, (33) demonstrating that this behavior is not unique to a single data set.
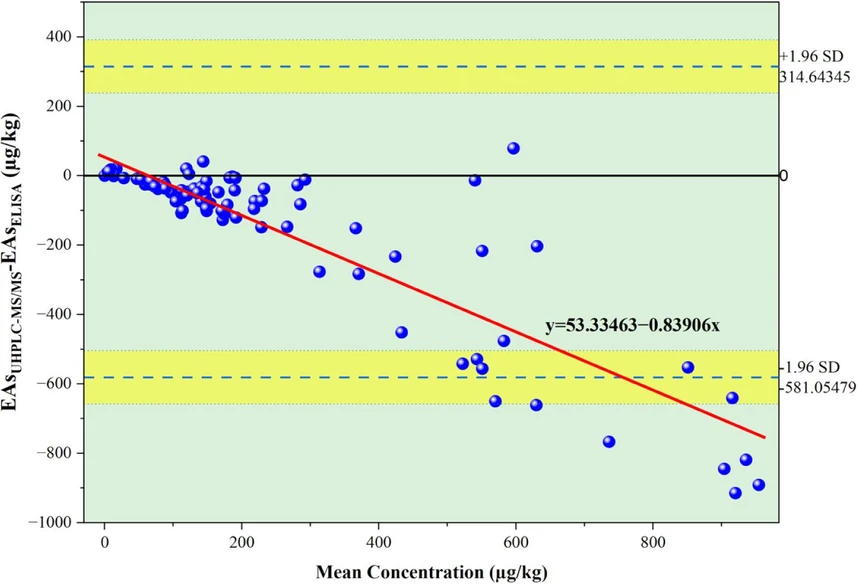 J. Agric. Food Chem. 2026, 74, 25, 20015–20027: Figure 6. Bland-Altman plot comparing ELISA and UHPLC-MS/MS measurements of total ergot alkaloids for wheat and durum wheat cargo samples (n = 102).
J. Agric. Food Chem. 2026, 74, 25, 20015–20027: Figure 6. Bland-Altman plot comparing ELISA and UHPLC-MS/MS measurements of total ergot alkaloids for wheat and durum wheat cargo samples (n = 102).
This study provides a comprehensive evaluation of a commercially available ELISA for total ergot alkaloids, using wheat samples as the primary application matrix, and integrates performance metrics with mechanistic insights into method behavior. While the assay demonstrated strong repeatability, calibration stability, and robustness to procedural factors such as test portion mass and extraction conditions, systematic differences relative to UHPLC-MS/MS were consistently observed. By linking these differences to compound-specific antibody cross-reactivity and matrix-dependent signal modulation, this work clarifies that ELISA-derived results reflect an immunochemically weighted response rather than a stoichiometric total. This distinction is critical for the appropriate interpretation of ELISA data and supports its use within tiered analytical strategies as a rapid screening tool, complemented by UHPLC-MS/MS for confirmatory quantification and regulatory decision-making.