Elucidating Unknown Organofluorine in Municipal Wastewater: A Mass Balance Approach including Fluorinated Pharmaceuticals
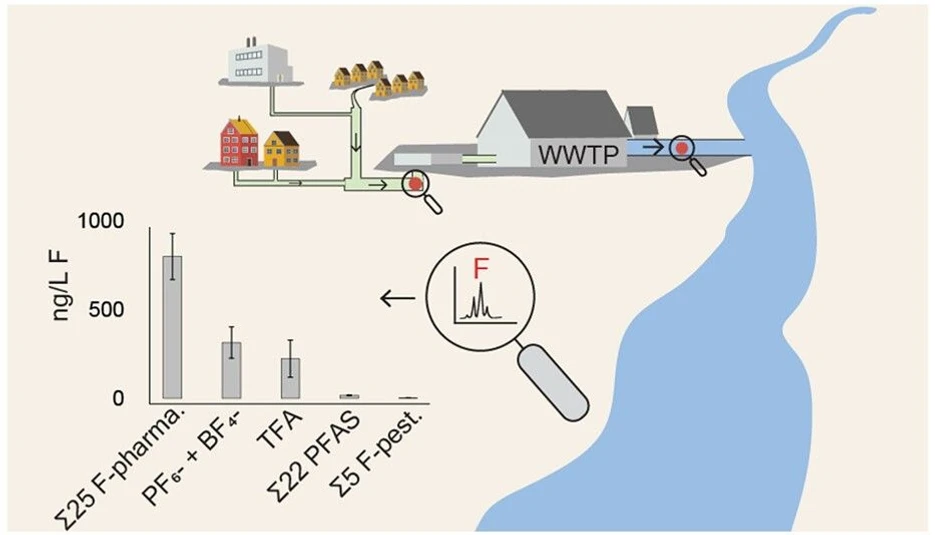
Environ. Sci. Technol. 2026, 60, 8, 6623–6634: Graphical abstract
This study investigates unidentified organofluorine in municipal wastewater using a multisorbent solid-phase extraction method combined with LC-HRMS screening and fluorine mass balance analysis. Highly fluorinated PFAS were detected at low concentrations, while numerous low-fluorinated pharmaceuticals and transformation products occurred at much higher levels.
Despite lower fluorine content, these pharmaceuticals accounted for a substantial share of extractable organofluorine, highlighting their importance in wastewater fluorine budgets. The results demonstrate the complexity of fluorine mass balance and emphasize the need for advanced analytical workflows to identify unknown fluorinated contaminants.
The original article
Elucidating Unknown Organofluorine in Municipal Wastewater: A Mass Balance Approach including Fluorinated Pharmaceuticals
Pontus Larsson, Anna Kärrman, and Leo W.Y. Yeung*
Environ. Sci. Technol. 2026, 60, 8, 6623–6634
https://doi.org/10.1021/acs.est.5c13161
licensed under CC-BY 4.0
Selected sections from the article follow. Formats and hyperlinks were adapted from the original.
Fluorine occurs naturally almost exclusively in inorganic forms, (1) but rare examples of naturally occurring organofluorines exist─for instance, certain plants produce organic monofluorinated compounds like fluoroacetate. Conversely, synthetic organofluorine has been extensively manufactured since the 1940s. Due to the unique properties of fluorine, organofluorines have found diverse applications─e.g., in refrigerants, (2) nonstick coatings, (3) water- and oil-repellant products, (4) pharmaceuticals, (5) and agrochemicals. (6) Per- and polyfluoroalkyl substances (PFAS), a subgroup of organofluorines, have gained increased interest due to their widespread occurrence in the environment and biota, (7,8) as well as their adverse health effects on humans and wildlife. (9) The chemical diversity within the PFAS subgroup is large. The total number of PFAS depends on the chemical definition and relevancy, but estimates range from ∼250 commercially relevant PFAS (10) to over 7 million possible structures. (11) Certain fluorinated pharmaceuticals and agrochemicals (e.g., fluoxetine, an antidepressant, and diflufenican, an herbicide), as well as refrigerants (e.g., R-134a), all of which contain at least one C–CF3 moetity, are considered PFAS according to a definition published in 2021 by the Organization for Economic Co-operation and Development (OECD). (12) However, according to certain organizations and governments, the above-mentioned compounds fall outside the PFAS subgroup. (13) Herein, the term conventional PFAS (14) is used to broadly refer to perfluoroalkyl acids (PFAA) and their precursors, while excluding compounds containing a C–CF3 moiety, such as certain pharmaceuticals and agrochemicals that may be considered precursors to the shortest-chain perfluorinated carboxylic acid (PFCA), trifluoroacetic acid (TFA).
The applicability of analytical methods suitable for determining PFAS as a group, e.g., assessing the total PFAS content in drinking water, (15) has been discussed. (16,17) Methods evaluated have included, among others: 1) high-resolution mass spectrometry (HRMS)-suspect screening and nontargeted workflows; (18) 2) sample oxidation techniques to reveal unknown precursors; (19,20) and 3) the use of combustion ion chromatography (CIC) to measure extractable (organo)fluorine (EOF/EF). The latter has previously been applied to various matrices, including wastewater. (21−23) EOF methods are commonly combined with targeted liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis to compare the fraction of organofluorine quantified by targeted approaches with the broader, nonspecific coverage of the CIC method. This concept is often referred to as fluorine mass balance analysis (24) and is used to estimate the amount of organofluorine unaccounted for in targeted analyses. Previous research (22) has indicated that a large fraction of the EOF cannot be explained by conventional PFAS, resulting in a significant proportion of unknown EOF. Recently, it was shown that fluorinated pharmaceuticals can account for a substantial fraction of previously unidentified EOF in municipal wastewater and sludge. (14,25) Previous fluorine mass balance studies of wastewater have exclusively employed a hydrophilic–lipophilic balance (HLB) or weak anion exchange (WAX) (22−24,26) sorbent for the solid phase extraction (SPE) of wastewater samples, which intrinsically targets neutral and anionic species. The use of multifunctional sorbents and multilayered SPE has been investigated previously to enhance the retention and extraction recovery of a broader range of analytes (27−29) but has yet to be applied to EOF methods. An assessment of a more inclusive extraction method for EOF analysis (e.g., multisorbent SPE) is, therefore, warranted.
This study introduces a workflow to improve the assessment of EOF in municipal wastewater by employing a multisorbent SPE to capture a broader spectrum of analytes. The workflow also enhances the quantification of organofluorine detected via HRMS screening, providing a more accurate evaluation of organofluorine loads entering and exiting wastewater treatment plants (WWTPs). By combining multisorbent SPE with advanced analytical techniques, this study elucidates the chemical composition of organofluorine and assesses the contributions of various fluorinated compounds, ranging from low-fluorinated substances, such as fluorinated pharmaceuticals and related compounds, to highly fluorinated conventional PFAS, to the overall organofluorine load in municipal wastewater.
2. Methods
2.3.2. LC-HRMS Screening
High-resolution accurate mass (HRAM) data were acquired using an LC (Acquity UPLC, Waters Corporation, Milford, USA) coupled to a quadrupole time-of-flight mass spectrometer (G2-XS, Waters Corporation, Milford, USA), operated in electrospray negative ionization (ESI−) and electrospray positive ionization (ESI+) modes in separate injections. The data were obtained using data-independent acquisition (MSe), rather than a data-dependent acquisition mode, to improve retrospective analysis capability. A low collision energy (3 eV) scan (denoted MS1) was followed by a high energy scan (denoted MS2), with a collision energy ramp from 10 to 35 eV. The scan range was m/z 50–1200 with a scan time of 0.2 s. Within-run mass calibration was performed by infusion of a solution containing leucine enkephalin every 10 s. UNIFI 1.9.4 (Waters Corporation, Milford, MA, USA) was used for data acquisition, lock-mass correction, and further peak processing. Details are referred to SI including Table S4.
Suspect screening using HRAM data was performed via curated lists of fluorinated pharmaceuticals and pesticides, including metabolites and transformation products, as well as conventional PFAS. Further information on suspect lists is provided in the SI. Feature prioritization was done by applying mass filtering at 5 ppm mass error and an isotope match filter (isotope match intensity RMS percent: 40 and isotope match m/z RMS ppm: 10). Further data curation included MS2 spectral matching using the MassBank online repository, (32) literature, and in silico fragmentation tools (UNIFI 1.9.4, MetFrag (33)), together with manual peak curation, e.g., fragment ion search. Reference materials for tentative suspects were acquired based on confidence in the identification and availability. Typically, the confidence level (CL) of identification reached CL2-3 (34) (i.e., most tentative candidates with MS2 fragment(s) matching with a library or diagnostic in silico fragments).
2.3.3.1. LC-MS/MS Conventional PFAS Analysis
The targeted analysis of conventional PFAS using LC-MS/MS has been described in detail and validated previously. (35) In short, an Acquity UPLC was coupled to an XEVO TQ-S tandem mass spectrometer (UPLC-MS/MS, Waters Corporation, Milford, MA, USA), operating in multiple reaction monitoring (MRM).
2.3.3.3. SFC-MS/MS Analysis of Polar Anions
For compounds exhibiting little to no retention on a reversed-phase liquid chromatography system, a supercritical fluid chromatograph (SFC) coupled to a tandem mass spectrometer (Acquity Ultra Performance Convergence Chromatograph, XEVO TQ-S micro, Waters Corporation, Milford, MA, USA) was used. Operating in ESI– mode and utilizing both MRM and single ion recording (SIR) for acquisition, five ultrashort-chain PFAAs (C2–C3 perfluorocarboxylic acids (PFCAs), C1–C3 perfluorosulfonic acids (PFSAs)), three inorganic fluorinated anions (hexafluorophosphate [PF6–]), tetrafluoroborate [BF4–] and bis(fluorosulfonyl)imide [FSI]), and one pharmaceutical (5-fluorouracil) were separated and analyzed in a single run.
2.3.4. Combustion Ion Chromatography for Extractable Organofluorine Analysis (CIC-EOF)
The CIC-EOF has been described elsewhere. (24) In brief, an aliquot (0.1 mL) of the sample extract was combusted at 1000–1050 °C, where the formed hydrogen fluoride (HF) was adsorbed in ultrapure water, separated, and quantified as fluoride in an ion chromatograph. An autosampler and combustion module (Analytik Jena, Jena, Germany) were used together with a 920 Absorber Module and 930 Compact IC Flex (Metrohm, Herisau, Switzerland). Further details on method evaluation are provided in SI and elsewhere. (36)
3. Results and Discussion
3.1. LC-HRMS-Based Screening
The focus of the LC-HRMS screening was to investigate the occurrence of low-fluorinated compounds (i.e., a fluorine mass fraction of ∼40% or less) not commonly targeted in PFAS analyses. Full-scan data were matched against a curated suspect list containing mainly fluorinated pharmaceuticals and metabolites. These matches were manually interrogated and constituted the basis for the acquisition of standards. Six compounds detected via suspect screening only reached a tentative identification level; these are summarized in Table 1 and are briefly discussed below. An expanded discussion on the HRMS data of tentatively detected compounds can be found in the SI and Table S12.
 Environ. Sci. Technol. 2026, 60, 8, 6623–6634: Table 1. List of Tentatively Identified Compounds in WWTP Influent and Effluent(a)
Environ. Sci. Technol. 2026, 60, 8, 6623–6634: Table 1. List of Tentatively Identified Compounds in WWTP Influent and Effluent(a)
A feature assigned as a hydrogen-substituted perfluoroether sulfonic acid (likely 1:2 H-PFESA) was tentatively detected in both the influent and effluent. This compound has previously been tentatively identified in wastewater (China (38) and Belgium (39)) and recently in a large European screening study. (40) No other hydrogen-substituted perfluoroether sulfonic acids (PFESAs) were detected in the present study, and no chlorine-substituted PFESAs were detected in the targeted analysis that may be precursors to certain H-PFESAs. 1:2 H-PFESA was identified at CL 2b and semiquantified to 1–10 ng L–1. Moreover, N-methylperfluorobutane sulfonamidoacetic acid (MeFBSAA) was identified at CL 2b and semiquantified to 1–10 ng L–1. Perfluoroalkane sulfonamidoacetic acids have been reported as intermediate environmental transformation products. (41)
3.3. Extractable (Organo)Fluorine Mass Balance
The EOF concentration ranged from 1300 to 3300 ng L–1 F (influent) and 1400–2300 ng L–1 F (effluent), which is comparable to earlier reported concentrations in Nordic WWTPs. (22) Conversion of mass concentration of individual compounds to fluorine equivalent concentrations (presented as ng L–1 F) was performed for all compounds (details are referred to SI). An overview of the fluorine mass balance is presented in Figure 2, including fluorine equivalent concentrations (panel A), EOF concentration and composition (panel B), distribution of the fluorine mass fraction, and fluorine equivalent concentrations in the effluent (panel C).
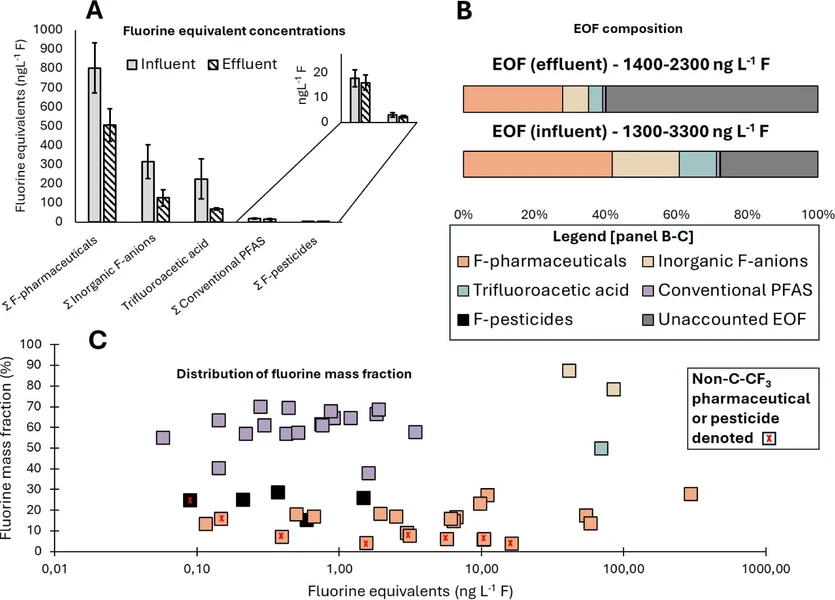 Environ. Sci. Technol. 2026, 60, 8, 6623–6634: Figure 2. Composition and concentration of extractable (organo)fluorine compounds in wastewater influent and effluent. (A) Fluorine equivalent concentrations (expressed as ng L–1 F); error bars display the standard deviation of the average (n = 3) of triplicate or duplicate extractions. (B) Average concentrations of compound groups (converted to ng L–1 F) in relation to the sum value of extractable (organo)fluorine, i.e., the EOF composition. (C) Distribution of quantified fluorinated compounds in effluent (converted to ng L–1 F) in relation to their fluorine mass fraction, calculated from the weight of fluorine atom(s) divided by the compounds’ molecular weight. Note: concentrations are not extraction recovery-corrected.
Environ. Sci. Technol. 2026, 60, 8, 6623–6634: Figure 2. Composition and concentration of extractable (organo)fluorine compounds in wastewater influent and effluent. (A) Fluorine equivalent concentrations (expressed as ng L–1 F); error bars display the standard deviation of the average (n = 3) of triplicate or duplicate extractions. (B) Average concentrations of compound groups (converted to ng L–1 F) in relation to the sum value of extractable (organo)fluorine, i.e., the EOF composition. (C) Distribution of quantified fluorinated compounds in effluent (converted to ng L–1 F) in relation to their fluorine mass fraction, calculated from the weight of fluorine atom(s) divided by the compounds’ molecular weight. Note: concentrations are not extraction recovery-corrected.
As shown in Figure 2, panels B and C, low-fluorinated organofluorine accounted for the majority of the EOF in influent. Most contained the substructure R-CF3, where R was a ring structure (e.g., aryl-CF3). This is in agreement with a recent study (68) where fluorine nuclear magnetic resonance (F-NMR) spectroscopy was used to identify aryl-CF3-containing compounds as the major organofluorine associated with samples from WWTPs. In the present study, fluorinated pharmaceuticals, including their metabolites, had the greatest sum of fluorine equivalents, making up, on average, 42% (influent) and 28% (effluent) of the EOF. Within this group, active pharmaceutical ingredients made up a larger proportion; 37% in influent and 23% in effluent, with metabolites thereof accounting for 5% in both influent and effluent. Fluorinated pesticides accounted for less than 0.2% of the EOF, indicating that they are not important drivers. TFA made up a large proportion of the EOF, accounting for 11% of the influent and 4% in effluent. Excluding TFA, conventional PFAS accounted for 1% in both the influent and effluent. This is lower than what has been reported (22) earlier Swedish effluent (average 14%), which may indicate a lower proportion of the EOF is explained by conventional PFAS in recent years. BF4– and PF6– together made up 19% and 7% of the EOF in influent and effluent, respectively, demonstrating the importance of inorganic fluoroanions in a fluorine mass balance context. In addition, due to the poor recovery of TFA and BF4–, it introduces a bias in the results that must be considered. Extraction recovery was greater in influent (TFA 22%, BF4– 39%) than effluent (TFA 10%, BF4– 17%); thus, TFA and BF4– concentrations in the extracts are not representative of the water sample and further lead to a higher fraction of the TFA and BF4– explaining the EOF in influent compared to effluent.
The most significant pharmaceuticals and related compounds contributing to the EOF were sitagliptin, followed by bicalutamide and celecoxib carboxylic acid, together accounting for 22–39% in influent and 19–26% in effluent. Sitagliptin alone accounted for an average of 29% in the influent and 16% in the effluent. In municipal wastewater from the United States, (25) sitagliptin accounted for 9–100% of the EOF in influent and 10–45% in effluent, and in suspended particulate matter from German rivers, (69) up to 94% of ∑8Organofluorine (detected via nontarget screening) was attributed to sitagliptin.
4. Environmental Implications and Future Needs
Previous studies have shown that wastewater treatment plants release organofluorine compounds into the environment, many of which remain unidentified. (22,23) Understanding their sources and quantifying the unidentified fraction are crucial to reducing human and environmental exposure. Persistent organofluorine may be released directly as stable compounds (e.g., PFAAs) or formed indirectly from certain substructures (e.g., C–CF3), which can degrade to TFA. This study advanced our knowledge by identifying low-fluorinated compounds, primarily pharmaceuticals and metabolites, as major contributors to EOF in wastewater. This is highlighted by several low-fluorinated compounds previously not quantified in municipal wastewater or the environment. Importantly, many of these compounds contained moieties with the potential to partly transform into TFA, whose concentrations in freshwater have increased rapidly in recent years. (47,48,72) Our results provide a clue to potential sources of TFA in the environment, but a better understanding of the fate and transformation of C–CF3 compounds in wastewater treatment systems is critical for accurate assessment.
In this study, conventional PFAS were not significant contributors to the EOF mass balance. Thus, the use of EOF-CIC for assessing PFAS totality in wastewater needs careful consideration for its purpose, given the high mass of fluorinated pharmaceuticals, including monofluorinated, entering municipal wastewater. Instead, improved strategies are needed, e.g., more specific sum determination of persistent organofluorine species. For example, Zweigle et al. (2024) introduced the concept of fractionation of low-fluorinated organofluorine and highly fluorinated PFAA precursors via oxidative conversion. With further development of this concept, EOF measurement of wastewater could enable a selective determination of persistent EOF (e.g., sum parameter determination of all hydroxyl-radical-stable perfluoroalkyls). Furthermore, a multisorbent SPE method enables extraction of a broader range of anionic, neutral, and cationic compounds, providing greater nonspecific coverage of the method. Improved sample cleanup methods that separate inorganic fluorides, including inorganic fluorinated substances (e.g., via initial hydrolysis of PF6– (73)) while quantitatively recovering small, polar organofluorines (e.g., TFA), would enable more unbiased and true EOF determination. These improvements could enable more accurate monitoring of persistent organofluorine in wastewater.